
Principal Investigator
The evolution of tumors is shaped by cell divisions and cell death. Our group is exploring the mechanisms underlying constitutive cancer cell death, in particular ferroptosis - a novel type of regulated necrosis.
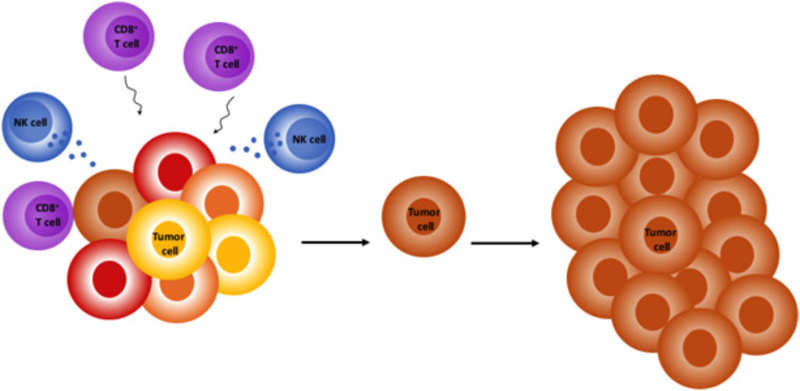
Cancers undergo “Darwinian” selection throughout their existence which may be triggered cell intrinsically, by immune surveillance or through therapy. While apoptosis resistance is a hallmark of cancer, several cancers were described to be instead sensitive to ferroptosis - an oxidative type of cell death. Our lab studies mechanisms of ferroptosis execution in cancer in order to unravel basic mechanisms, immunological consequences and potential therapeutic applications in human disease.
Our lab’s overarching aim is to understand which cell death pathways are selected against by the immune system during cancer development. By unravelling mechanisms of selection-of-the-fittest cellular clone, they anticipate a better understanding of what these cells are selected for and thereby identify new treatment opportunities for cancer.
To this end, our lab makes use of several genetically engineered mouse model systems including small cell lung cancer (SCLC), B-cell lymphoma and pancreatic ductal adenocarcinoma (PDAC).
In ongoing projects, our lab investigates the role of caspase 8 in selection-of-the-fittest in SCLC and PDAC, as well as metabolic ferroptosis vulnerability and regulation in SCLC subtypes and B-cell lymphoma.
Although its key pathway players are well conserved throughout phylogeny, ferroptosis has only been discovered in 2012. It seems we know the least about some of the most primitive types of cell death. But primitive is good, it means cancers will be vulnerable to these mechanisms.
Principal Investigator